

Abstract
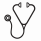
Background: Heat shock factor 1 (HSF1) is a key-component of the heat shock response and plays a major role in cancer biology. The heat shock response is a highly conserved mechanism that is important for cell survival under stressful conditions. It facilitates normal protein folding and guards the proteome from misfolding and aggregation. Bortezomib (BTZ) is a reversible proteasome inhibitor and was recently tested in a phase 3 clinical trial for patients with de novo pediatric acute myeloid leukemia (pedi-AML). This trial compared standard therapy (cytarabine/daunorubicin/etoposide (ADE)) to ADE plus BTZ (ADE+B). As HSF1 and BTZ act in opposing circumstances, our goal was to globally assess the phosphorylation of HSF1 at serine 326 (HSF1.pS326) and to determine if baseline HSF1.pS326 was prognostic of clinical response to ADE+B and if we could identify a subset of patients that benefitted from ADE+B.
Methods: Reverse phase protein array (RPPA) probed with 222 total antibodies and 69 specific post-translationally modified proteins was used to determine the protein expression levels in 505 bulk leukemic pedi-AML samples compared to 20 CD34+ samples from healthy pediatric donors. Patients participated in the Children's Oncology Group (COG) AAML1031 Phase 3 clinical trial that compared ADE to ADE+B. Survival curves were generated using the Kaplan-Meier method.
Results: HSF1.pS326 protein expression levels in the pedi-AML patient samples. Based on the HSF1.pS326 expression levels, patients were classified into two clusters; high HSF1 326 (n = 216) and low HSF1.326 (n = 289). The median of the normal CD34+ samples was used as cut-off point. Expression of HSF1.pS326 was not prognostic for outcome in patients treated with ADE (event-free survival (EFS), p = 0.51) (Fig. 1A). In patients that were treated with ADE+B however, HSF1.pS326 levels were highly prognostic, with low HSF1.pS326 associated with a better EFS (p = 0.009) (Fig. 1B). Patients with high HSF1.pS326 did not benefit from BTZ addition (EFS, p = 0.77) (Fig. 1C), whereas patients with low HSF1.pS326 levels significantly improved (EFS, p = 0.004) (Fig. 1D). Between the two patient clusters, the NPM1 mutation state was higher in patients with high HSF1.pS326 levels (p = 0.042), and the CEBPA mutation state was higher in patients with low HSF1.pS326 (p = 0.011). No other patient or disease characteristics were different between the two clusters.
To functionally validate our findings, we overexpressed wild type or mutant HSF1 in 293T cells and assessed its effect on BTZ sensitivity. The mutant HSF1 constructs contained amino acid substitutions at Ser326, resulting in either a phospho-mimic HSF1 (S326E), or a phospho-dead HSF1 protein (S326A). Cell viability analysis showed clear resistance to BTZ in cells with increased phosphorylation (HSF1 S326E) (Fig. 1E - blue curve). Cells that expressed reduced phosphorylation of HSF1 (HSF1 S326A), however were highly sensitive to BTZ (Fig. 1E, red curve). This confirms our observations from the pedi-AML patients.
Conclusions: Using a proteomics approach, we identified a subgroup of pedi-AML patients that benefitted from BTZ addition to ADE therapy. We hypothesize that patients with low HSF1 activation are more susceptible to protein cell stress, and protein cell stress susceptibility is amplified by the addition of BTZ with ADE. The use of HSF1.pS326 as potential biomarker could identify patients that would benefit from ADE+B therapy. Patients with high HSF1 could potentially benefit from ADE+B in combination with a HSF1 (phospho) inhibitor, suggesting a novel combinational therapy.
Figure 1. Kaplan Meier survival curves for EFS based on HSF1.pS326 expression. A. HSF1.pS326 did not affect outcome in ADE treated patients (high HSF1.PS326 shown in red, low HSF1.pS326 in blue). B. HSF1.pS326 was highly prognostic for EFS ADE+B. C. Patients with high HSF1.pS326 did not benefit from BTZ (ADE shown in blue, ADE+B in green). D. Patients that expressed low HSF1.pS326 did significantly better after ADE+B. E. To assess the effect of high and low HSF1.pS326 on BTZ sensitivity, 293T cells were transfected with two HSF1.pS326 mutants that mimicked high (blue) and low (red) HSF1.pS326 patients, in addition to wild type HSF1 (orange) and an empty vector as control (green). Cell viability assays showed resistance to BTZ in the phospho-mimic (blue) mutant, whereas the phospho-dead (red) mutant was sensitive to BTZ.
Kolb:Roche- Genentech: Membership on an entity's Board of Directors or advisory committees; Servier: Membership on an entity's Board of Directors or advisory committees.
This icon denotes a clinically relevant abstract

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal